再生医療等製品の製造において、細胞培養加工施設(CPC)のクリーンルーム管理は品質と安全性を担保する生命線です。しかし、施設の設計や運用基準を策定する際、工業規格である「ISO 14644-1」のクラス分類と、医薬品製造管理基準である「GMP」のグレード分類(A~D)の整合性に悩まれる管理者の方も多いのではないでしょうか。
本記事では、再生医療の現場で求められる「クリーンルームの清浄度クラス(ISO・GMP)」について、両者の定義の違いや換算方法、そして実務における運用ポイントを専門的な視点から解説します。適切な環境モニタリング基準を確立し、査察対応や汚染リスク低減にお役立てください。
再生医療におけるクリーンルーム清浄度クラス(ISO・GMP)の対応と結論
再生医療の製造現場において、クリーンルームの清浄度管理は極めて重要です。しかし、ISO規格とGMPガイドラインでは、その目的や定義に微妙な差異が存在します。ここではまず、両者の基本的な違いと、実務上でどのように対応させるべきか、その結論となる換算関係について解説します。
ISO 14644-1規格とGMPグレード(A~D)の定義の違い
ISO 14644-1規格とGMPグレード(A~D)は、いずれも清浄度を示す指標ですが、その着眼点が異なります。ISO規格は主に「気中の微粒子(パーティクル)数」に基づき、物理的な清浄度をクラス1から9まで分類する工業的な統一規格です。
一方、GMP(特にPIC/S GMPやEU-GMP Annex 1)におけるグレードA~Dは、微粒子数に加えて「微生物汚染のリスク管理」を重視しています。再生医療等製品の製造においては、単に空気がきれいなだけでなく、製品への微生物混入を防ぐための無菌操作環境が求められるため、ISOの数値をベースにしつつ、GMPのグレード要件を満たすという二重の視点が必要となります。
非作業時(At-rest)と作業時(In-operation)における基準の使い分け
GMPグレードの大きな特徴として、「非作業時(At-rest)」と「作業時(In-operation)」という2つの状態を区別して基準を設けている点が挙げられます。
- 非作業時(At-rest): 設備が稼働し、人がいない状態
- 作業時(In-operation): 設備が稼働し、作業員が通常の作業を行っている状態
ISO規格では通常、特定の状態を指定して測定しますが、GMPではこの状態変化による汚染リスクの変動を考慮します。特にグレードB区域では、非作業時にはグレードAと同等の清浄度が求められますが、作業時には許容される微粒子数が増加します。この「状態による基準の使い分け」こそが、ISO単独の管理では不十分な理由です。
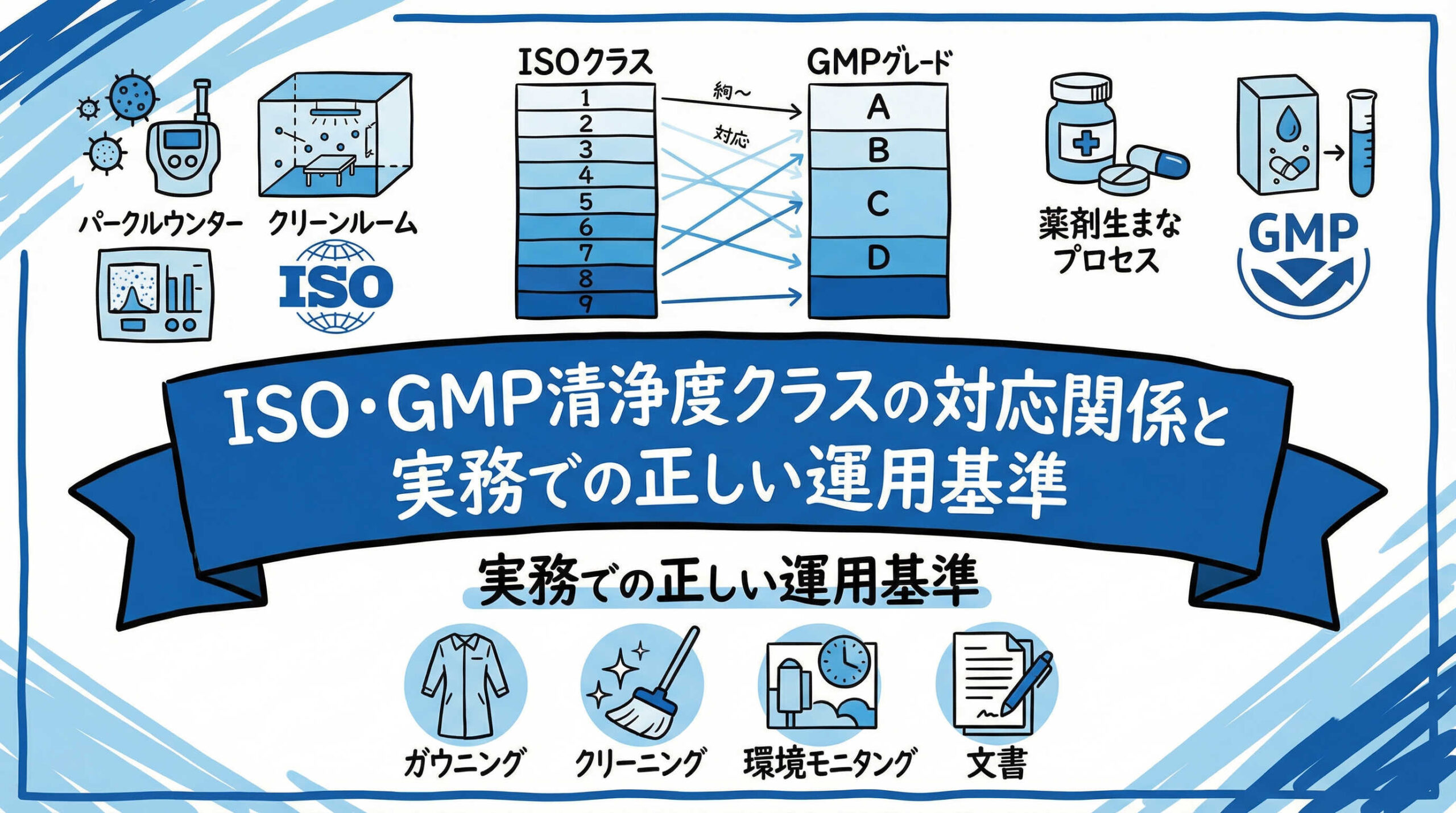
ISOクラスとGMPグレードの換算・対応関係一覧表
実務において最も頻繁に参照されるのが、ISOクラスとGMPグレードの対応関係です。以下の表は、一般的な目安としての換算・対応関係を示したものです。
| GMPグレード | 定義(用途) | ISOクラス(非作業時) | ISOクラス(作業時) |
|---|---|---|---|
| グレードA | 重要区域(無菌操作) | ISO 4.8 / ISO 5 | ISO 5 |
| グレードB | グレードAの背景区域 | ISO 5 | ISO 7 |
| グレードC | 清浄区域 | ISO 7 | ISO 8 |
| グレードD | 前室・補助区域 | ISO 8 | 規定なし |
このように、グレードBは「非作業時はISO 5、作業時はISO 7」というように変動するため、設計段階からこの要件を満たす空調能力や運用ルールを考慮する必要があります。
再生医療の現場でISOとGMPの基準を厳格に管理する法的根拠と背景
再生医療等製品は、最終製品の滅菌が困難であるため、製造工程全体を通じた無菌性の保証が不可欠です。ここでは、なぜISOとGMPの基準を厳格に管理する必要があるのか、その法的根拠と背景にあるリスク管理の考え方について掘り下げていきます。
GCTP省令およびGMP省令における構造設備の要件
再生医療等製品の製造管理および品質管理の基準に関する省令(GCTP省令)では、構造設備に対して製品の品質を確保するために必要な清浄度を求めています。GCTPは医薬品のGMP省令をベースにしていますが、細胞などの特性を考慮した柔軟性も一部認められています。
しかし、構造設備の要件としては、汚染や交叉汚染(クロスコンタミネーション)を防止するための適切な空調システムの設置や、作業室の清浄度区分ごとの明確な分離が必須です。これらの法的要件を満たすためには、単なる感覚的な管理ではなく、ISOやGMPに基づいた客観的なデータによる裏付けが求められるのです。
無菌操作法指針(無菌医薬品製造)に基づく環境管理の重要性
日本国内において、再生医療等製品の製造環境を整備する際に重要なガイドラインとなるのが「無菌操作法指針」です。この指針では、無菌医薬品の製造と同様に、作業区域をグレードA~Dに分類し、それぞれの区域で許容される微粒子数や微生物数の基準を示しています。
特に再生医療の現場では、細胞の培養や加工といった開放系での操作(オープンハンドリング)が発生する場面があります。このような重要工程においては、無菌操作法指針に基づき、グレードA環境下での作業を徹底し、その背景環境であるグレードB区域の管理も厳格に行うことが、製品の安全性を担保する上で極めて重要です。
細胞培養加工施設(CPC)における汚染リスクと清浄度の関係
細胞培養加工施設(CPC)における最大のリスクは、細菌、真菌、ウイルス、マイコプラズマなどによる生物学的汚染です。一般的な医薬品と異なり、生きた細胞を扱う再生医療製品は、最終工程での加熱滅菌やフィルター濾過ができないケースがほとんどです。
清浄度クラスの維持は、これらの汚染物質が製品に混入する確率を限りなくゼロに近づけるための物理的な障壁となります。ISO規格に基づく微粒子管理は、微生物が付着する「足場(パーティクル)」を減らすことで間接的に微生物汚染を防ぎ、GMPに基づく微生物モニタリングは直接的な汚染リスクを監視します。この両輪が機能して初めて、安全な製品製造が可能となります。
GMPグレード(A・B・C・D)ごとの詳細要件とISOクラス換算
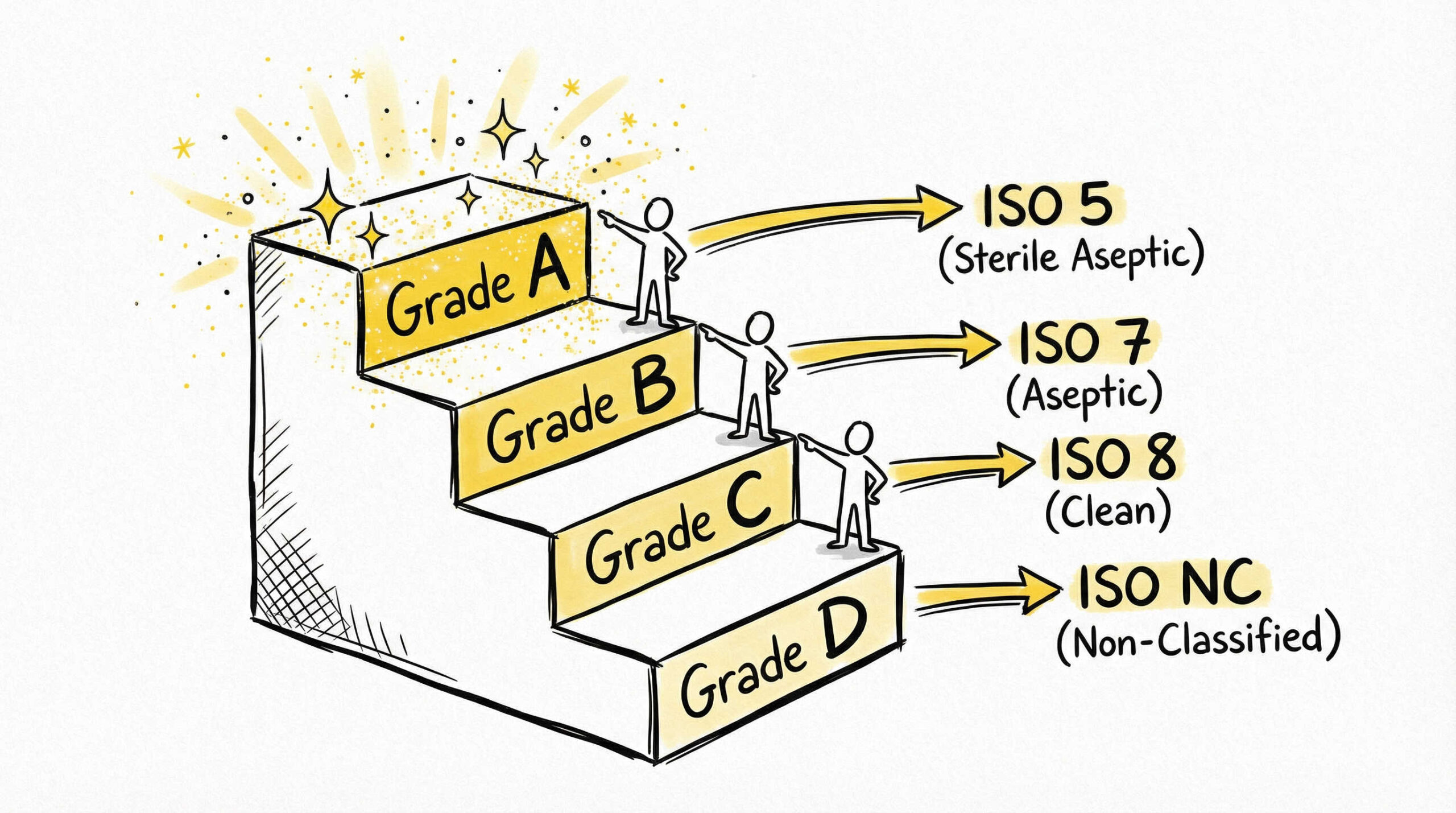
各GMPグレードには、微粒子数だけでなく微生物の許容菌数など、詳細な要件が定められています。ここでは、現場担当者が押さえておくべきグレードAからDまでの具体的な基準と、それに対応するISOクラスの詳細について解説します。
グレードA(ISO 4.8 / ISO 5相当):重要区域・無菌操作部の基準
グレードAは、製品が直接外気に触れる無菌操作が行われる最も重要な区域です。具体的には、生物学的安全キャビネット(BSC)やクリーンベンチの内部がこれに該当します。
- ISO換算: ISO 4.8(5.0μm以上の微粒子を考慮する場合)またはISO 5に相当。
- 特徴: 作業時・非作業時を問わず、気中微粒子数は極めて低いレベルに維持される必要があります。また、一方向流(ラミナーフロー)によって清浄空気が供給され、汚染物質を速やかに排除できる構造が求められます。微生物に関しては、検出限界以下(<1 CFU)を目指す厳格な管理が必要です。
グレードB(ISO 5 / ISO 7相当):グレードAの直接支援区域の基準
グレードBは、グレードA区域(キャビネット内など)を取り囲む背景環境であり、無菌操作を行うための直接的な支援区域です。作業員が実際に活動する部屋そのものが該当します。
- ISO換算: 非作業時はISO 5、作業時はISO 7に相当。
- 特徴: 非作業時にはグレードAと同等の清浄度への回復能力が求められます(リカバリーテスト)。作業中は人の動きにより発塵が増えますが、それでもISO 7レベルを維持し、グレードAへの汚染持ち込みを防ぐバッファとしての役割を果たします。ここでの更衣管理や動線管理が、グレードAの品質を左右すると言っても過言ではありません。
グレードC(ISO 7 / ISO 8相当):その他の清浄区域の基準
グレードCは、無菌操作工程ほど重要度は高くないものの、清浄度が管理された区域です。溶液の調製や、最終的な無菌ろ過を行う前の工程などで使用されます。
- ISO換算: 非作業時はISO 7、作業時はISO 8に相当。
- 特徴: 再生医療の現場では、培養室へ入る前の廊下や、細胞調製室の前室などがこのグレードに設定されることが一般的です。グレードBエリアへ入室する前の段階として、一定レベルの清浄度を維持し、外部からの汚染負荷を低減させる役割を担います。モニタリングの頻度はグレードA・Bに比べて緩和されますが、定期的な監視は必須です。
グレードD(ISO 8相当):作業区域の前室および補助区域の基準
グレードDは、製造区域としての最低限の清浄度が定義された区域です。一般区域(CNC:Controlled Not Classified)と清浄区域の境界に位置します。
- ISO換算: 非作業時はISO 8に相当。作業時の規定は明確にはありませんが、管理状態にあることが求められます。
- 特徴: 一次更衣室や、洗浄後の器具の保管場所、部材の搬入室などが該当します。ここでは、外部からの汚染を遮断し、より高いグレードの区域へ汚染を持ち込まないための基本的な衛生管理が行われます。靴の履き替えや手洗い、一次ガウニングなどがこの区域の主な管理ポイントとなります。
適合性確認のための環境モニタリングと清浄度測定方法

クリーンルームが規定の清浄度(ISO・GMP)を満たしているかを証明するためには、継続的な環境モニタリングが必要です。ここでは、適合性確認のために実施すべき具体的な測定手順と、判定基準の考え方について解説します。
気中微粒子(パーティクル)数の測定手順と判定基準
気中微粒子数の測定には、校正されたパーティクルカウンター(微粒子計測器)を使用します。測定は、ISO 14644-1に基づき、部屋の面積に応じたサンプリング点数を決定して行います。
- 測定対象: 主に0.5μm以上と5.0μm以上の粒子径。
- 判定基準: 各グレード(A~D)および状態(非作業時・作業時)ごとに定められた上限値を超えないこと。
特にグレードAでは、連続的なモニタリングシステムを用いて、作業中の突発的な発塵イベントを常時監視することが推奨されます。データは記録として残し、トレンド分析を行うことで異常の予兆を捉えることが重要です。
浮遊菌・落下菌・表面付着菌の微生物モニタリング手法
微粒子測定だけでは見えない「生物学的汚染」を監視するのが微生物モニタリングです。以下の手法を組み合わせて実施します。
- 浮遊菌測定: エアーサンプラーで空気を吸引し、培地に捕集して培養。
- 落下菌測定: 開放した寒天培地(落下菌測定用プレート)を一定時間設置し、落下してくる菌を捕集。
- 表面付着菌測定: コンタクトプレート(スタンプ法)やスワブ法を用いて、壁、床、機器表面、作業員の手指などを検査。
これらの測定結果はCFU(Colony Forming Unit)で評価され、各グレードの許容菌数以下であることが求められます。菌が検出された場合は、菌種の同定を行い、汚染源を特定する必要があります。
差圧管理と気流方向(エアフロー)の確認方法
清浄度の高い区域から低い区域への空気の流れ(エアフロー)を維持し、汚染の逆流を防ぐために「室圧(差圧)」の管理は不可欠です。
- 差圧: 隣接するグレードの異なる部屋間で、通常10〜15Pa以上の差圧を確保します(陽圧管理)。
- 気流方向: スモークテスターなどを用いて気流を可視化し、空気が清浄区域から汚染側へ適切に流れているか、乱流や滞留がないかを確認します。
差圧計は日常的に目視確認するほか、警報システムと連動させ、扉の開放などで差圧が低下した際に即座に対応できる体制を整えましょう。
環境モニタリングの実施頻度とサンプリングポイントの設定
モニタリングの頻度や場所は、リスクアセスメントに基づいて決定します。一律に決めるのではなく、「汚染リスクが高い場所」や「製品品質に直結する場所」を重点的に監視します。
- グレードA: 製造作業中、継続的に実施(特に粒子モニタリング)。
- グレードB: 作業シフトごとに実施するなど、高頻度での監視。
- グレードC/D: 週次や月次など、定期的な確認で傾向管理を行う。
サンプリングポイントは、作業員の動線、機器の近く、給気口・排気口の位置などを考慮して設定し、SOP(標準作業手順書)に明確に記載しておきましょう。
CPC(細胞培養加工施設)における清浄度維持と運用管理のポイント
クリーンルームの設備性能だけでなく、日々の運用管理(ソフト面)が清浄度維持の鍵を握ります。CPC(細胞培養加工施設)における具体的な運用ルールやメンテナンスのポイントについて詳しく見ていきましょう。
清浄度区分に応じた更衣(ガウニング)手順と入退室管理
クリーンルーム内での最大の発塵源は「人」です。そのため、グレードに応じた適切な更衣(ガウニング)手順の遵守が不可欠です。
- グレードA/B: 無菌衣、フード、マスク、滅菌手袋、滅菌ブーツなどを着用し、皮膚の露出を極限まで減らします。着衣手順は検証され、認定された作業員のみが入室できるようにします。
- 入退室管理: インターロック付きのパスルームを使用し、一度に複数の扉を開けないこと、更衣室での動線(汚染側と清浄側の分離)を守ることなどを徹底します。
定期的な更衣評価(ガウニング適格性評価)を行い、作業員の手技レベルを維持することも重要です。
施設内の清掃・消毒(サニテーション)プログラムの策定
施設内の清浄度を保つためには、科学的根拠に基づいた清掃・消毒(サニテーション)プログラムが必要です。
- 消毒剤の選定: 複数の消毒剤(アルコール系、次亜塩素酸系、過酢酸系など)をローテーションで使用し、耐性菌の出現を防ぎます。
- 芽胞菌対策: 定期的に殺芽胞剤(スポリサイド)を使用し、アルコールでは死滅しない芽胞菌のリスクを管理します。
- 手順の標準化: 「上から下へ」「奥から手前へ」といった清拭の基本ルールをSOP化し、拭き残しがないように教育します。
清掃用具自体も滅菌済みのものや、発塵の少ない専用品を使用しましょう。
HEPAフィルタのリークテストと定期的な性能適格性評価
空調システムの要であるHEPAフィルタは、定期的な性能確認が義務付けられています。
- リークテスト(完全性試験): フィルタおよび取付け枠からの漏れがないか、PAOなどの試験粒子を用いて検査します。通常、年1回以上の実施が推奨されます。
- 性能適格性評価: 風速、風量、換気回数が設計値を満たしているか、清浄度回復時間が適切かを定期的に再検証(リバリデーション)します。
これらの定期点検により、フィルタの目詰まりや破損による性能低下を早期に発見し、予期せぬ汚染事故を未然に防ぐことができます。
米国連邦規格(Fed-Std-209D/E)の旧クラス表記との関係性
古い文献や施設の図面では、米国連邦規格「Fed-Std-209D/E」に基づく「クラス100」「クラス10,000」といった表記を見かけることがあります。この規格自体は既に廃止されていますが、現場では慣習的に使われることがあるため、ISOクラスとの関係を知っておくと便利です。
- クラス100 ≒ ISO 5 (グレードA/B相当)
- クラス10,000 ≒ ISO 7 (グレードC相当)
- クラス100,000 ≒ ISO 8 (グレードD相当)
あくまで目安ですが、この対応関係を理解しておくと、既存施設の改修や海外の古い資料を参照する際に役立ちます。
まとめ
本記事では、再生医療におけるクリーンルームの清浄度クラスについて、ISO規格とGMPグレードの対応関係を中心に解説してきました。
重要なポイントは以下の通りです。
- ISOとGMPの違い: ISOは微粒子数、GMPはそれに加えて微生物管理と「作業時・非作業時」の状態区分がある。
- 換算の目安: グレードAはISO 5、グレードBは非作業時ISO 5/作業時ISO 7、グレードCはISO 7/8、グレードDはISO 8に概ね相当する。
- 運用の重要性: 設備(ハード)だけでなく、モニタリング、更衣、清掃といった運用(ソフト)が清浄度維持には不可欠。
CPCの管理者やQA/QC担当者は、単に数値をクリアするだけでなく、その背景にある「汚染リスクの低減」という目的を常に意識し、自施設に最適な管理基準を構築していくことが求められます。
クリーンルームの清浄度クラス(ISO・GMP)解説についてよくある質問

以下に、再生医療分野のクリーンルーム管理において頻繁に寄せられる質問とその回答をまとめました。実務の参考としてご活用ください。
- Q1. ISOクラスとGMPグレードの最も大きな違いは何ですか?
- ISOクラスは主に「微粒子数」で分類する工業規格ですが、GMPグレードは微粒子に加え「微生物汚染リスク」や「作業状態(作業時/非作業時)」を考慮した医薬品製造のための管理基準です。
- Q2. グレードAとグレードBの境界はどのように管理すべきですか?
- 物理的な隔壁(キャビネットのガラス面など)に加え、気流(エアフロー)によるエアカーテン効果で管理します。また、作業員の手指がグレードAエリアに入る際は厳格な消毒が必要です。
- Q3. 再生医療の現場で特に注意すべき汚染源は何ですか?
- 最大の汚染源は「作業員(人)」です。皮膚からの発塵や呼気中の微生物が主なリスクとなるため、更衣(ガウニング)の徹底と動作の適正化が最も効果的な対策となります。
- Q4. 環境モニタリングの頻度はどのように決めればよいですか?
- リスクアセスメントに基づき決定します。無菌操作を行うグレードA/Bは作業ごとの実施が基本ですが、トレンドデータが安定していれば頻度を調整できる場合もあります。無菌操作法指針などを参考にSOPを策定しましょう。
- Q5. 昔の図面に「クラス100」とありますが、現在の基準ではどれに該当しますか?
- 米国連邦規格(Fed-Std-209E)の「クラス100」は、概ね「ISO 5」およびGMPの「グレードA/B(非作業時)」に相当します。現在は廃止された規格ですが、現場用語として残っている場合があります。