再生医療等製品の製造において、クリーンルームの品質管理は製品の安全性と有効性を担保する生命線です。特にGCTP省令への適合を目指す品質保証担当者様にとって、適切な「クリーンルームのバリデーションと環境モニタリング」の実施計画策定は、避けて通れない重要な課題ではないでしょうか。
施設の立ち上げ時や定期的な査察対応において、規制要件を満たしたバリデーションの手順や、日常的な環境モニタリングの基準値を明確に把握しておくことは不可欠です。本記事では、再生医療の現場に即した具体的かつ実用的なバリデーションの実施手順、測定項目、そして環境モニタリングの運用ポイントについて、専門的な視点からわかりやすく解説します。貴社の品質管理体制をより強固なものにするための一助として、ぜひお役立てください。
クリーンルームのバリデーションと環境モニタリングはGCTP適合の要
再生医療におけるクリーンルーム管理において、「バリデーション」と「環境モニタリング」は車の両輪のような関係にあります。これらは混同されがちですが、GCTP(再生医療等製品の製造管理及び品質管理の基準)に適合するためには、それぞれの目的と役割を明確に区別し、かつ有機的に連携させることが重要です。ここでは、両者の基本的な違いと、無菌性保証における位置づけについて解説します。
バリデーションと環境モニタリングの違いと相互関係
バリデーションとは、クリーンルームの設備やシステムが「期待される性能を恒常的に発揮できること」を検証し、文書化するプロセスです。いわば、その設備が製造に適格であることを証明する「スナップショット」のようなものです。
一方、環境モニタリングは、バリデーションされた状態が日常の製造過程において「維持されていること」を継続的に監視・測定する活動を指します。バリデーションで適格性を確認し、環境モニタリングでその維持管理を確認するという相互補完的な関係性を理解しましょう。
再生医療等製品の製造における無菌性保証の考え方
再生医療等製品は、最終製品に対する無菌試験だけでは品質を完全に保証することが困難なケースが多々あります。そのため、製造プロセス全体を通じた「無菌性保証」の考え方が極めて重要となります。
クリーンルームのバリデーションと環境モニタリングは、このプロセス保証の中核を担います。単に数値をクリアするだけでなく、科学的根拠に基づいた管理によって、製品への汚染リスクを最小限に抑える環境が構築されていることを論理的に説明できる体制が求められるのです。
バリデーション(適格性評価)を実施する目的と規制要件

クリーンルームのバリデーションを実施することは、単なる自主管理ではなく、GCTP省令をはじめとする法規制によって求められる義務でもあります。なぜバリデーションが必要なのか、どのような基準で実施すべきか、その背景にある規制要件と目的を深く理解することで、より効果的な品質管理計画を策定できるでしょう。ここでは、主要な規制要件とリスク管理の視点について掘り下げます。
GCTP省令における構造設備への要求事項
GCTP省令では、製造所の構造設備が製品の品質に悪影響を及ぼさないよう、適切に設計、配置、および保守されることが求められています。具体的には、清浄度区分に応じた空調システムの設置や、交差汚染を防止するための動線管理などが含まれます。
バリデーションは、これらの構造設備が設計通りの性能を有し、規制要件を満たしていることを客観的なデータで実証するために不可欠なプロセスです。査察時には、これらの記録が適合性の証拠として重視されます。
汚染管理戦略(CCS)に基づいたリスク評価の重要性
近年のGMP/GCTPのトレンドでは、汚染管理戦略(CCS: Contamination Control Strategy)の策定が強く推奨されています。これは、施設全体のリスクを包括的に評価し、汚染を防ぐための戦略を文書化するものです。
バリデーション計画も、このCCSに基づいて策定されるべきです。一律の基準を適用するだけでなく、製品の特性やプロセスの重要度に応じたリスク評価を行い、リスクが高い箇所にはより厳格な検証を行うといったメリハリのある管理が求められます。
定期バリデーションと変更時の再バリデーション
バリデーションは一度実施すれば終わりではありません。経年劣化や環境変化に対応するため、定期的な再バリデーション(通常は1年に1回程度)が必要です。
また、空調設備の改造や製造プロセスの変更、あるいは環境モニタリングで逸脱が続いた場合などには、変更時の再バリデーションを実施しなければなりません。常に最新の状態が適格であることを保証し続けるサイクルを回すことが、品質保証の信頼性を高めます。
クリーンルームの適格性評価(バリデーション)の具体的な実施手順
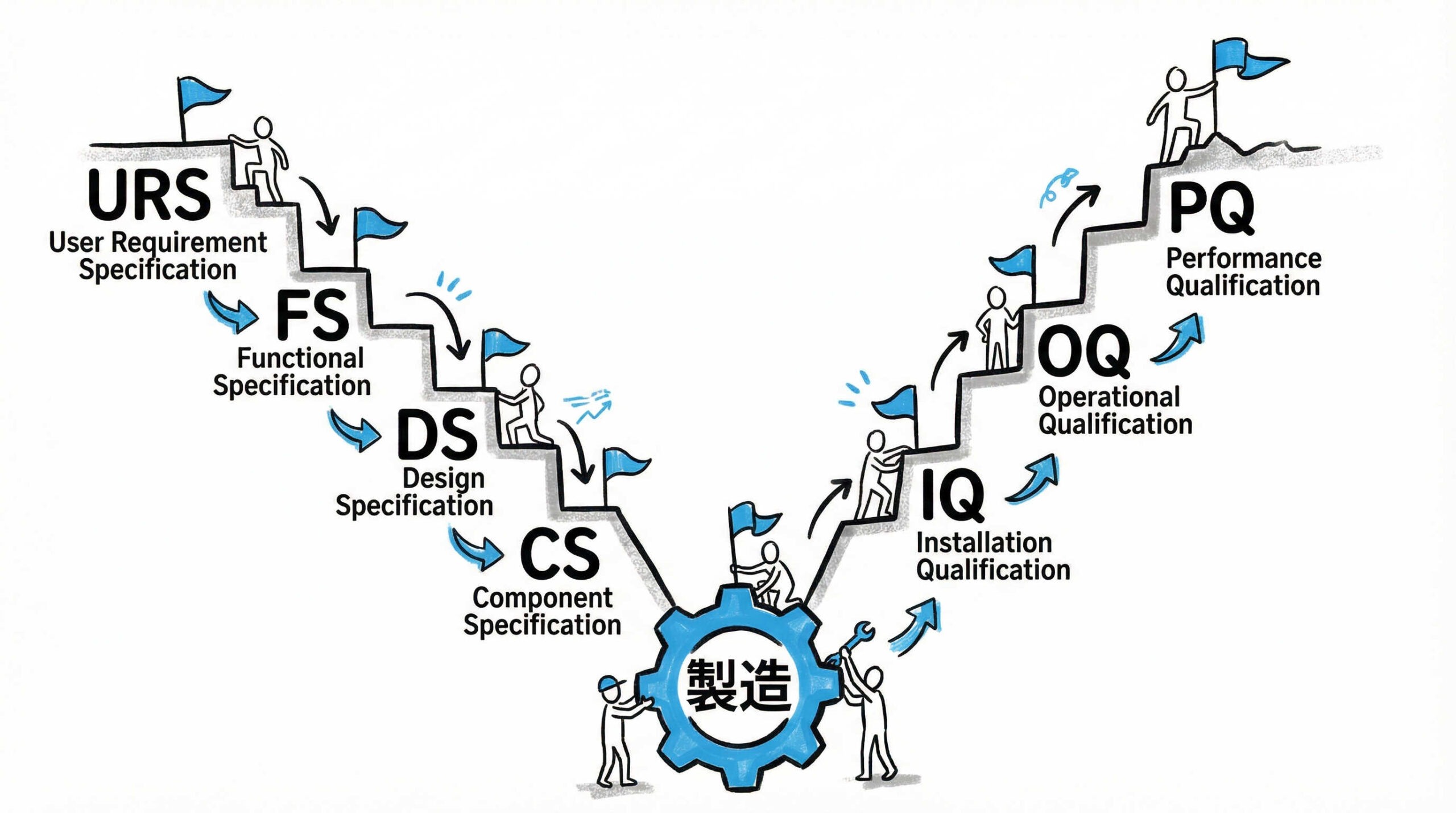
クリーンルームの適格性評価(バリデーション)は、一般的に「Vモデル」と呼ばれる流れに沿って段階的に進められます。計画段階から実際の稼働状態に至るまで、各フェーズで何を確認すべきかを明確にすることが成功の鍵です。ここでは、URSからPQに至るまでの具体的な実施手順を、実務的な視点で解説します。
要求仕様書(URS)と設計時適格性評価(DQ)
すべての出発点は、要求仕様書(URS)の作成です。ここでは、製造する製品の特性に合わせて、必要な清浄度クラス、温湿度条件、差圧設定などを明確に定義します。
続いて、設計時適格性評価(DQ)を行います。これは、設計図面や仕様書がURSの要件を満たしているかを机上で検証するプロセスです。施工前に設計上の不備を見つけ出し、手戻りを防ぐための重要なステップと言えるでしょう。
設備の据付時適格性評価(IQ)
設備の据付時適格性評価(IQ)は、実際に施工された設備や機器が、設計図書やメーカーの仕様通りに正しく設置されているかを確認する検証です。
具体的には、主要機器の型番確認、配管やダクトの接続状況、計器類の校正状況(キャリブレーション)、材質の確認などを実施します。この段階で施工ミスや部材の間違いがないことを確実にし、次の運転確認へと進みます。
設備の運転時適格性評価(OQ)
運転時適格性評価(OQ)では、設備を実際に稼働させ(通常は無負荷状態)、設計された通りの性能が発揮されるかを確認します。
空調機の風量、室圧制御、温度・湿度の制御範囲、警報システムの動作確認などが含まれます。この段階では、製造機器などを稼働させない状態で、クリーンルームという「器」としての基本性能が満たされているかを検証します。
実際の製造環境を想定した性能適格性評価(PQ)
最後の仕上げとなるのが、性能適格性評価(PQ)です。これは、実際に製造機器を稼働させ、作業員が在室するなど、実際の製造環境を模擬した状態(負荷状態)で性能を検証するプロセスです。
製造プロセスと同様の条件下でも、清浄度や温湿度が規定の範囲内に維持されるかを確認します。PQのデータは、その後の環境モニタリングの基準設定の根拠ともなるため、非常に重要な意味を持ちます。
バリデーションにおける主要な測定・検証項目
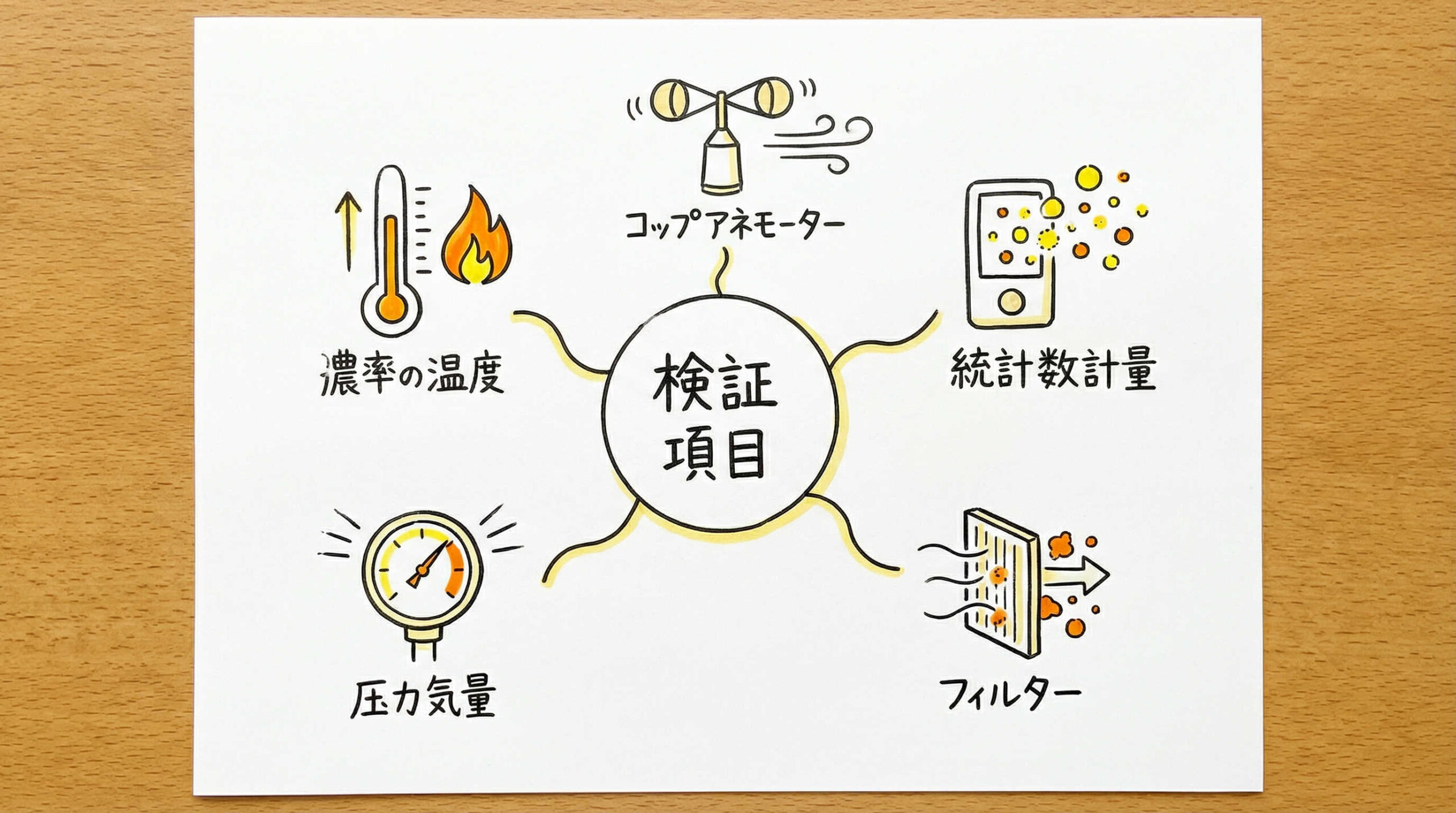
バリデーションの実務において、具体的にどのような項目を測定し、何を検証するのかを知ることは極めて重要です。これらの測定項目は、JIS B 9920やISO 14644などの規格に基づいて実施されます。ここでは、クリーンルームの性能を評価する上で特に重要となる5つの測定・検証項目について詳しく解説します。
HEPAフィルタの完全性試験(リーク試験)
クリーンルームの清浄度を担保する要となるのがHEPAフィルタです。完全性試験では、フィルタの濾材や枠のシール部分に漏れ(リーク)がないかを確認します。
一般的には、PAOなどの試験粒子を上流側から供給し、下流側(室内側)で粒子をスキャンして漏れを検知します。わずかなピンホールでも汚染の原因となるため、定期的な実施が欠かせません。この試験に合格することが、清浄度確保の第一歩です。
風速・風量および換気回数の測定
清浄度を維持するためには、十分な換気回数が必要です。各吹出口からの風速と風量を測定し、部屋全体の換気回数を算出します。
グレードに応じた適切な換気回数が確保されているかを確認するとともに、風速の均一性(ユニフォーミティ)も評価します。特に一方向流(ラミナーフロー)方式の場合は、風速の乱れが汚染拡散につながるため、厳密な測定が求められます。
室間差圧の測定と気流方向の確認
クリーンルームでは、清浄度の高い部屋から低い部屋へと空気が流れるよう、室間に適切な圧力差(差圧)を設ける必要があります。これにより、外部からの汚染物質の流入を防ぎます。
バリデーションでは、各室間の差圧が設計値通りに維持されているか、そしてドアの開閉時にも気流の逆流が起きないかを確認します。適切な室間差圧の維持は、交差汚染防止の要です。
気流可視化試験(スモークテスト)
数値データだけでなく、実際の空気の流れを目で見て確認するのが気流可視化試験(スモークテスト)です。純水ミストやスモークテスターを用いて、気流の方向や挙動を観察します。
特に重要なのは、作業面での気流が適切か、機器周辺で空気が滞留(淀み)していないかを確認することです。滞留箇所は汚染物質が蓄積しやすいため、この試験結果をもとに作業手順や機器配置を見直すこともあります。
清浄度回復試験
清浄度回復試験は、万が一汚染が発生した場合に、空調システムがどれほどの速さで元の清浄度まで回復できる能力があるかを検証するものです。
人工的に粒子を発生させて清浄度を悪化させ、その後、規定の清浄度レベル(例えば100分の1の濃度)に戻るまでの時間を測定します。この「回復時間」を知ることは、汚染トラブル発生時の製造再開判断において重要な指標となります。
環境モニタリングの測定項目と管理基準値
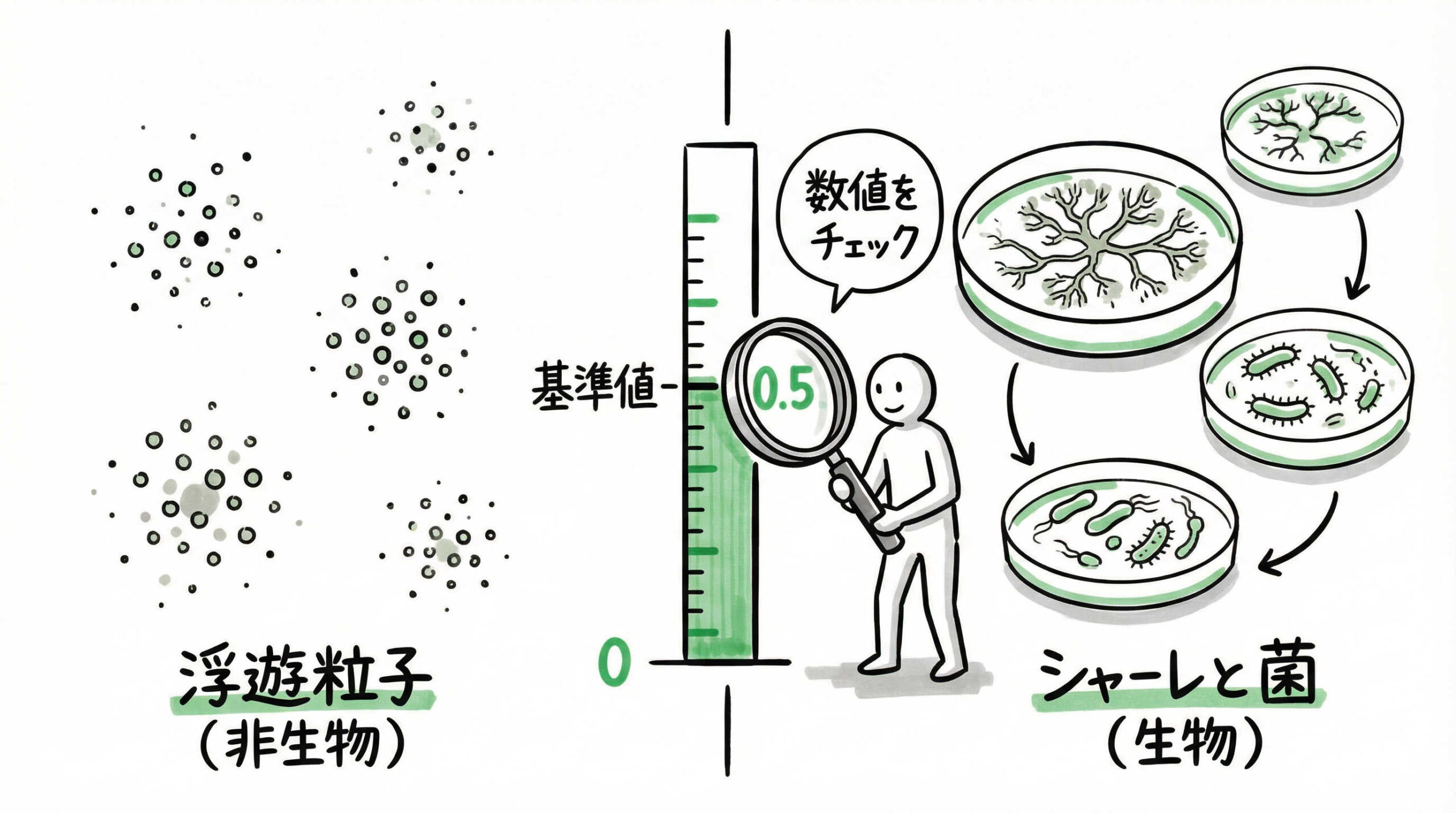
バリデーションで性能が証明された後は、日常的な環境モニタリングによってその状態を維持管理します。モニタリングには「非生物学的モニタリング」と「生物学的モニタリング」があり、それぞれに管理基準値の設定が必要です。ここでは、主要な測定項目と、再生医療の現場で求められる一般的な管理基準について解説します。
浮遊微粒子数(パーティクル)の測定基準
浮遊微粒子(パーティクル)の測定は、清浄度クラスの維持を確認する基本です。パーティクルカウンターを使用し、0.5μmおよび5.0μmの粒子数を測定します。
基準値はISO 14644-1やPIC/S GMP Annex 1などを参照して設定します。グレードAでは連続的なモニタリングが推奨される一方、グレードB以下ではリスクに応じた頻度で測定を行います。微粒子の増加は、フィルター破損や発塵源の存在を示唆する重要なサインです。
浮遊菌の測定方法と許容基準
浮遊菌の測定は、空気中に浮遊している微生物を捕集して培養する方法です。エアーサンプラーを用いて一定量の空気を寒天培地に吹き付け、培養後にコロニー数(CFU)をカウントします。
無菌操作を行うグレードA区域では、検出限界(<1 CFU/m³)が基準となることが一般的です。目に見えない微生物汚染を監視するため、サンプリングの位置やタイミングは、作業の影響を最も受けやすい場所を選ぶことが肝要です。
落下菌の測定方法と許容基準
落下菌測定は、寒天培地を一定時間開放し、重力によって落下してくる微生物や粒子に付着した微生物を捕集する方法です。
これは、作業中の製品や設備への直接的な汚染リスクを評価するのに適しています。通常、作業が行われている時間帯(最長4時間程度)に実施します。作業者の動きや気流の乱れによる影響を反映しやすいため、作業環境の評価として非常に有効です。
表面付着菌(スタンプ法・スワブ法)の測定
製造機器の表面、作業台、壁、床などの微生物汚染を確認するために、スタンプ法(フードスタンプ)やスワブ法(綿棒による拭き取り)を用います。
平滑な面にはスタンプ法が便利ですが、複雑な形状の箇所や隙間にはスワブ法が適しています。特に、製品が直接触れる可能性のある表面や、作業者が頻繁に触れる箇所は重点的にモニタリングし、清掃・消毒の効果を検証する必要があります。
5本指グローブによる手指付着菌モニタリング
再生医療における汚染源の多くは「人」に由来します。そのため、作業者の手袋(グローブ)の表面付着菌モニタリングは極めて重要です。
作業終了時や重要な工程の区切りに、5本の指先すべてを寒天培地に押し付けて測定します。グレードA/Bエリアでの作業後は必須となる項目であり、手指衛生の手順が遵守されているかを客観的に評価する指標となります。
温度・湿度の管理幅設定
温度と湿度は、作業者の快適性だけでなく、微生物の増殖抑制や静電気防止の観点からも管理が必要です。
一般的に、湿度は高すぎるとカビや細菌の増殖リスクが高まり、低すぎると静電気による微粒子の付着リスクが生じます。製品の安定性データや作業環境の特性に合わせて、適切な管理幅(例:温度23±5℃、湿度50±10%など)を設定し、連続的に記録・監視します。
再生医療におけるグレード別清浄度基準(A/B/C/D)
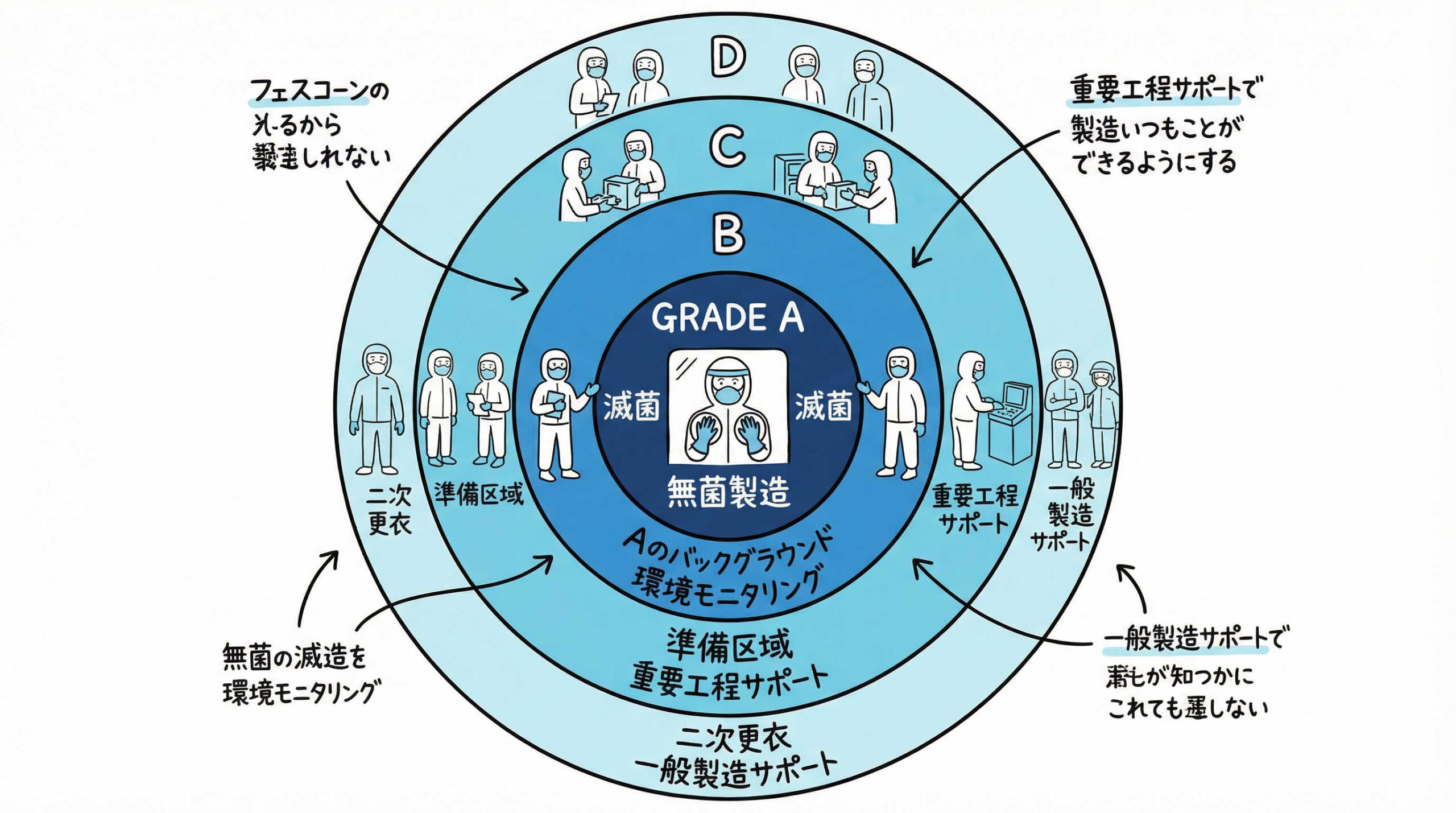
再生医療等製品の製造エリアは、その清浄度レベルに応じてグレードA、B、C、Dの4段階に区分されます。この区分けは、PIC/S GMP Annex 1などの国際的なガイドラインに基づいています。各グレードで求められる清浄度基準と管理レベルを正しく理解し、適切なゾーニングを行うことが汚染防止の基本です。
グレードA(重要区域)の基準と管理
グレードAは、製品の無菌操作や充填などを行う最も重要な区域であり、最高レベルの清浄度が求められます。一方向流(ラミナーフロー)によって空気が制御され、浮遊微粒子や微生物が限りなくゼロに近い状態を維持しなければなりません。
ここでは、作業中の環境モニタリングも連続的かつ厳格に行われ、作業者の介入も最小限に制限されます。アイソレータや安全キャビネット内部がこれに該当します。
グレードB(直接支援区域)の基準と管理
グレードBは、グレードAの直接的な背景となる区域です。無菌調製室などがこれに当たります。グレードAの清浄度を維持するためのバッファーゾーンとしての役割を果たします。
非作業時(At rest)にはグレードAと同等の微粒子清浄度が求められますが、作業時(In operation)には一定の許容範囲が設けられます。更衣や物品の搬入出における厳格な管理が、グレードBの維持には不可欠です。
グレードC・D(その他の支援区域)の基準と管理
グレードCおよびDは、製造プロセスの重要度が比較的低い工程(溶液の調製や器具の洗浄など)が行われる区域です。
グレードA/Bに比べて基準は緩やかですが、外部からの汚染を持ち込ませないための段階的な清浄化エリアとして機能します。これらのエリアでの適切な管理が、結果として重要区域の清浄度を守ることにつながるため、決して軽視してはいけません。
非作業時(At rest)と作業時(In operation)の区別
清浄度基準を理解する上で重要なのが、「非作業時(At rest)」と「作業時(In operation)」の区別です。非作業時は設備は稼働しているが人がいない状態、作業時は実際に製造活動が行われている状態を指します。
特に微粒子数に関しては、非作業時と作業時で基準値が大きく異なります。バリデーションやモニタリングでは、どの状態での測定値なのかを明確にし、それぞれの基準に適合しているかを評価する必要があります。
モニタリング計画の策定と運用のポイント
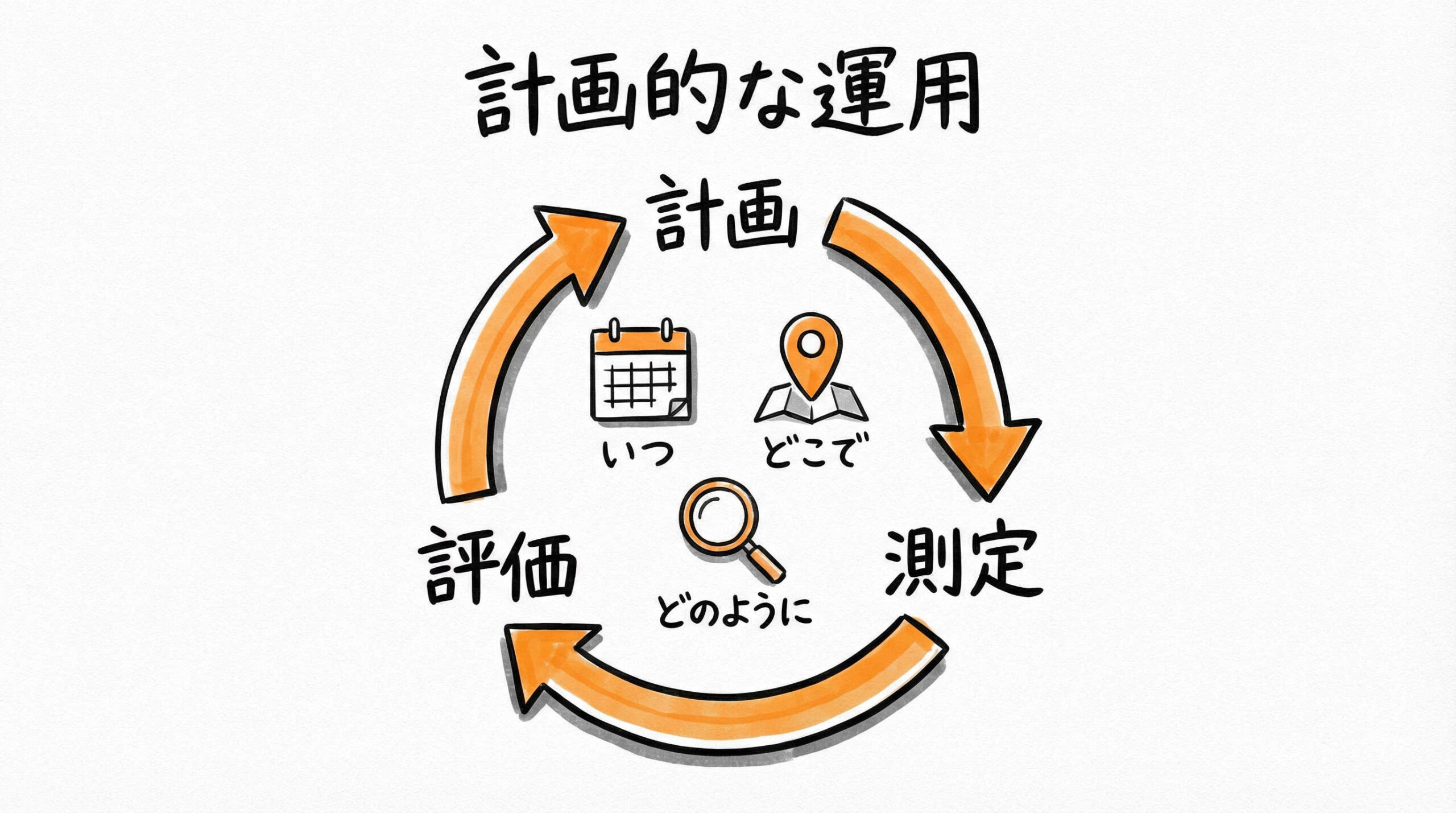
効果的な環境モニタリングを実施するためには、単に測定するだけでなく、計画的な運用体制の構築が不可欠です。「どこを」「いつ」「どのように」測定し、異常時にどう対応するか。ここでは、リスクベースアプローチに基づいた計画策定と、データの信頼性を確保するための運用のポイントについて解説します。
リスクアセスメントに基づくサンプリングポイントの選定
サンプリングポイントは、闇雲に決めるのではなく、リスクアセスメントに基づいて選定します。気流の滞留しやすい場所、作業者の動線が交差する場所、製品が曝露される場所など、汚染リスクが高い箇所を特定しましょう。
気流可視化試験(スモークテスト)の結果なども参考に、ワーストケース(最も条件の悪い場所)を含めることで、モニタリングの有効性が高まります。根拠のあるポイント選定は、査察時の説明責任を果たす上でも重要です。
測定頻度の決定プロセス
測定頻度は、製造する製品のリスクや工程の重要度に応じて決定します。グレードA/Bのような重要区域では、製造ロットごと、あるいはシフトごとの頻繁な測定が求められます。
一方、グレードC/Dでは週1回や月1回など、リスクに応じた頻度設定が可能です。ただし、過去のモニタリングデータの傾向分析(トレンド分析)を行い、安定している場合は頻度を見直すなど、柔軟かつ科学的な運用が推奨されます。
警報基準(アラートレベル)と処置基準(アクションレベル)の設定
モニタリングデータには、単なる合格/不合格の基準だけでなく、予兆管理のための「警報基準(アラートレベル)」と「処置基準(アクションレベル)」を設定します。
アラートレベルは、通常の変動範囲を超えたが規格内である状態で、環境悪化の兆候を捉えるためのものです。アクションレベルは、許容限度を超えた状態で、直ちに原因調査と是正措置が必要です。これらを適切に設定することで、深刻な汚染トラブルを未然に防ぐことができます。
データインテグリティ(DI)を考慮した記録管理
医薬品業界全体で重要視されているデータインテグリティ(DI:データの完全性)は、環境モニタリングにおいても適用されます。測定データは正確に記録され、改ざんが防止され、追跡可能でなければなりません。
手書き記録のダブルチェックや、電子記録システムの導入、監査証跡(オーディットトレイル)の管理など、データの信頼性を担保する仕組み作りが必要です。「記録がない=実施していない」とみなされることを肝に銘じましょう。
逸脱発生時のCAPA(是正予防措置)フロー
万が一、モニタリング結果が基準を逸脱した場合には、CAPA(是正予防措置)プロセスを回します。まずは応急処置を行い、次に根本原因を調査(Root Cause Analysis)します。
単に「再清掃して再測定」で終わらせるのではなく、なぜ逸脱が起きたのか(人の動き、空調不具合、清掃手順の不備など)を突き止め、再発防止策を講じることが重要です。このCAPAの履歴は、品質システムの成熟度を示す重要な証拠となります。
まとめ

本記事では、再生医療等製品の製造におけるクリーンルームのバリデーションと環境モニタリングについて、規制要件から具体的な実施手順までを解説してきました。
GCTP省令に適合し、安全な製品を患者様に届けるためには、バリデーションによる「適格性の証明」と、環境モニタリングによる「状態の監視」の両輪が機能することが不可欠です。リスクに基づいた科学的な管理計画を策定し、継続的な改善サイクル(CAPA)を回していくことが、貴社の品質保証体制をより盤石なものにするでしょう。専門業者への委託も含め、自社に最適な運用体制を構築してください。
クリーンルームのバリデーションと環境モニタリングについてよくある質問

ここでは、クリーンルームのバリデーションや環境モニタリングの実務において、品質管理担当者の方からよく寄せられる質問とその回答をまとめました。
- バリデーションの実施頻度はどのくらいが適切ですか?
- 定期的な再バリデーションは、一般的に1年に1回の頻度で実施することが推奨されます。ただし、リスクアセスメントの結果や施設の経年状況、環境モニタリングのトレンド分析結果に基づいて、頻度を調整することもあります。また、設備の大規模な変更や修繕を行った際は、その都度「変更時のバリデーション」が必要です。
- 環境モニタリングで菌が検出された場合、どう対応すべきですか?
- まずは検出された菌が「アラートレベル」か「アクションレベル」かを確認します。アクションレベルを超えた場合は、直ちに製造への影響評価を行い、原因調査(菌の同定など)と是正措置(清掃・消毒の強化など)を実施します。アラートレベルの場合も、傾向を監視し、頻発するようであれば予防的な措置を講じることが重要です。
- グレードAエリアでのモニタリングは、作業中ずっと行う必要がありますか?
- はい、原則としてグレードAエリアでの重要工程(無菌操作や充填など)の間は、浮遊微粒子等の連続的なモニタリングが求められます。これにより、作業中の突発的な環境悪化をリアルタイムで検知し、製品へのリスクを即座に把握することが可能になります。
- アラートレベルとアクションレベルの基準値はどのように決めますか?
- アクションレベルはガイドライン等の規制値を上限として設定します。一方、アラートレベルは自施設の過去のモニタリングデータを統計的に分析し、通常の変動範囲(例えば平均値+2σや3σなど)を基に設定するのが一般的です。これにより、自施設特有の異常の兆候を早期に捉えることができます。
- バリデーションや測定業務を外部委託する際の注意点は?
- 委託業者が適切な資格や経験を持っているか、使用する測定機器が校正(キャリブレーション)されているかを必ず確認してください。また、測定方法や判定基準が貴社の品質管理基準(URSなど)と整合しているかを事前にすり合わせ、最終的な報告書のレビューを自社の責任で行うことが重要です。